
We are developing a portfolio of GeneTAC® product candidates designed to address genetic diseases driven by inherited nucleotide repeat expansions. GeneTAC® molecules are designed to be a novel class of disease-modifying small molecule therapeutic candidates that can either dial up or dial down the expression of a specific disease-causing gene to address the underlying cause of disease.
Our proprietary GeneTAC® platform offers significant potential advantages over other modalities. When systemically administered, GeneTAC® molecules can distribute widely to reach target cells, overcoming a central challenge for traditional genomic medicines. GeneTAC® molecules are also designed to work with the natural genome without altering a patient’s DNA.
We are advancing four programs in severe monogenic diseases. Each of these programs has the potential to generate blockbuster products with first-in-class or best-in-class profiles.

Friedreich Ataxia Program
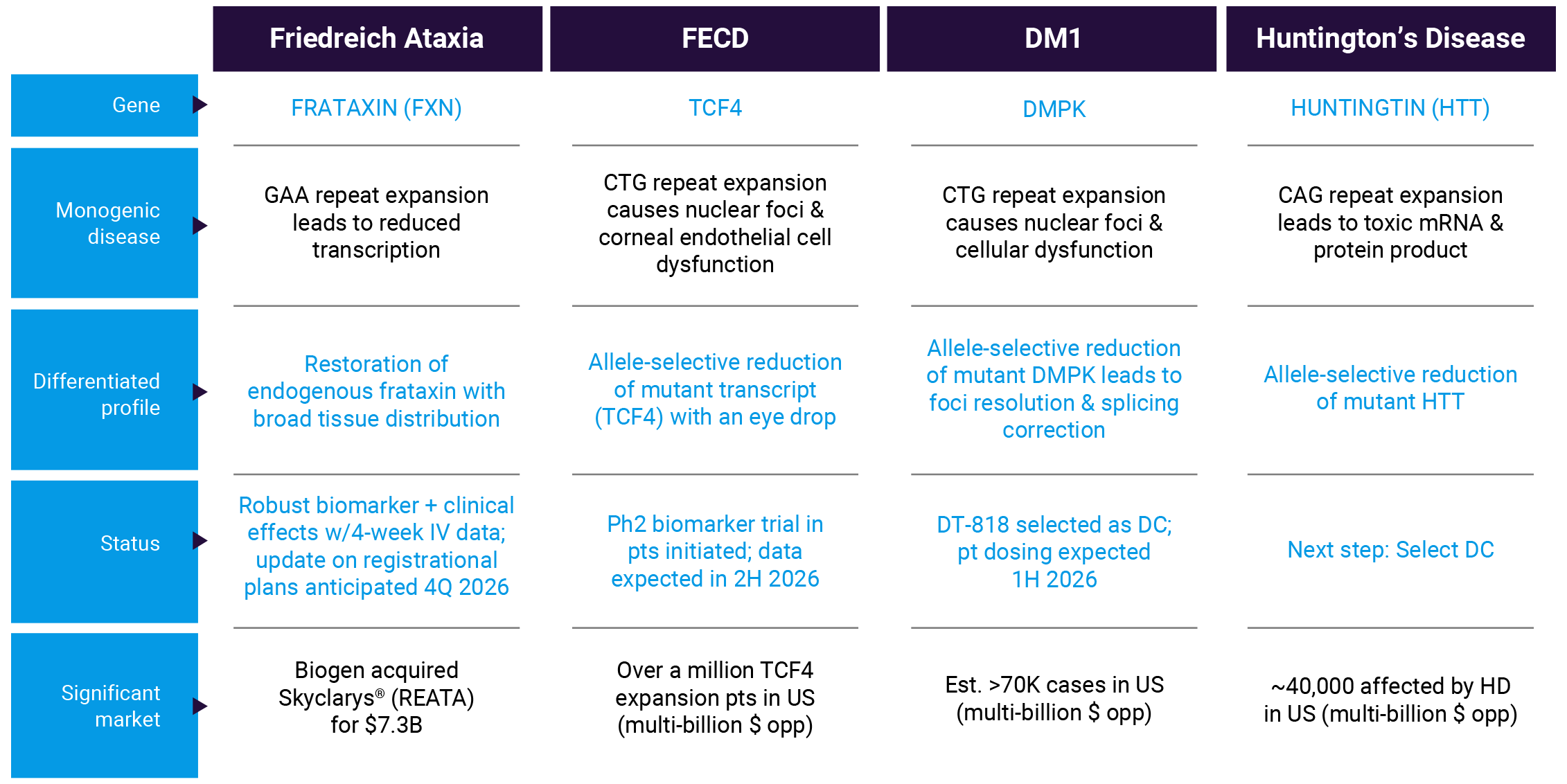
Friedreich ataxia (FA) is a devastating monogenic, autosomal recessive progressive disease caused by low levels of endogenous frataxin (FXN) due to abnormally expanded GAA triplet repeat expansions in the first intron of the FXN gene. The disease is characterized by spinocerebellar ataxia, dysarthria, pyramidal weakness, deep sensory loss, hypertrophic cardiomyopathy, skeletal abnormalities and diabetes mellitus. Clinical onset occurs most often around puberty, leading to severe disability by early adulthood, with substantial functional loss, wheelchair dependence and loss of quality of life. Affected individuals have reduced life expectancy, with many premature deaths caused by complications of cardiomyopathy at about the end of the fourth decade of life. The estimated prevalence of FA is 1 in 40,000–50,000, affecting more than 5,000 individuals living in the United States and more than 20,000 in Europe.
Our FA program, designed to address the underlying cause of disease, is based on GeneTAC® small molecules consisting of a DNA-binding moiety designed to bind to the expanded GAA sequence in the first intron of the FXN gene in FA patients, linked to a ligand moiety designed to recruit an endogenous transcriptional elongation complex to unblock the transcriptional machinery and restore the production of endogenous FXN proteins to therapeutic levels.
We are currently running a Phase 1/2 multiple dose trial of DT-216P2 in patients with FA known as RESTORE-FA (Reactivating Expression Suppressed Through Overcoming Repeat Expansion for FA). In May 2026, we announced four-week IV data from the RESTORE-FA Trial of DT-216P2 demonstrating dose-dependent improvement in multiple clinical measures and increases in endogenous frataxin mRNA and protein after four weeks of intravenous dosing. DT-216P2 was generally well-tolerated, with no serious adverse events or treatment discontinuations reported. Based on these data, we intend to pursue a registrational path and provide an update on our plans in the fourth quarter of 2026.
Fuchs Endothelial Corneal Dystrophy Program
Fuchs Endothelial Corneal Dystrophy (FECD) is characterized by degeneration of corneal endothelial cells (CECs) and progressive loss of vision. Typically, the disease manifests after age 40 and can be detected through routine eye exams. As individuals age, CECs become dysfunctional and degenerate and eventually fluid accumulates in the cornea (corneal edema). As disease progresses, FECD leads to reduced visual acuity, reduced contrast sensitivity, an increase in glare, and eventual corneal blindness. Other symptoms include pain and grittiness in the eye.
This eye disease affects millions of people worldwide. Approximately 60-80% of FECD cases are caused by cytosine-thymine-guanine (CTG) nucleotide repeat expansions in the TCF4 gene, which is transcribed into pathogenic TCF4 RNA that forms nuclear foci and sequesters splicing proteins, leading to transcript mis-splicing (spliceopathy) and loss of CECs. CECs harbor the longest known TCF4 repeat expansions in the body, potentially explaining why the cornea is the only affected tissue. There is currently no effective therapeutic intervention that addresses the root cause of the disease. Various modalities of keratoplasty, including corneal transplantation, constitute the only treatment option to correct FECD.
Our FECD program leverages our expertise in designing GeneTAC® small molecules that address the underlying cause of the disease. FECD GeneTAC® molecules we designed have been shown to markedly reduce nuclear foci and improve spliceopathy in FECD CEC cultures derived from the corneal tissue of donors who underwent corneal transplant.
DT-168 is a GeneTAC® small molecule designed to target the CTG repeats in the TCF4 gene and selectively block transcription of the expansion-containing allele. It is also designed to be applied as an eye drop.
We reported favorable results from the Phase 1 SAD/MAD clinical trial of DT-168 in healthy volunteers at Eyecelerator @ Park City 2025. The results demonstrated that DT-168 was well-tolerated with no serious adverse events, no ocular adverse events (AEs) and no treatment discontinuations due to AEs in the trial. All observed AEs were deemed not related to DT-168 by the investigator. Design has opened a Phase 2 biomarker trial of DT-168 to evaluate safety, tolerability and corneal endothelium biomarkers in FECD patients who are scheduled for corneal transplant surgery.
Myotonic Dystrophy Program
Myotonic dystrophy (DM1) is a monogenic, autosomal dominant, progressive neuromuscular disease that affects skeletal muscle, heart, brain and other organs. The cardinal features include muscle weakness, myotonia (slow muscle relaxation) and early cataracts. In addition, affected individuals often experience cardiac arrhythmias and changes in neuropsychological function. DM1 is caused by a mutation in the DMPK gene and is estimated to have a genetic prevalence of 1 in 2,300–8,000 people, affecting more than 70,000 people in the United States.
Our DM1 program is based on GeneTAC® small molecule candidates consisting of a DNA-binding moiety designed to target the CTG repeats in the 3’ untranslated region of the DMPK gene, linked to a ligand moiety that is designed to dial down transcription of the mutant expanded CTG repeat without disrupting the normal DMPK expression. As a result, the DM1 GeneTAC® molecules are designed to prevent the formation of the CUG hairpin structures that trap splicing proteins and produce nuclear foci. As with our other programs, the DM1 program is designed to address the underlying cause of the disease and benefit from the favorable development advantages of small molecules.
DT-818 is a GeneTAC® small molecule designed to target the CTG repeats in the DMPK gene and selectively reduce transcription of the mutant expanded allele.
We anticipate starting to dose DM1 patients in a Phase 1 multiple-ascending dose (MAD) clinical trial of DT-818 in Australia in the first half of 2026 to assess safety and correction of mis-splicing.
Huntington’s Disease Program
Huntington’s disease (HD) is a dominantly inherited, monogenic neurodegenerative disease characterized by progressive movement, cognitive and psychiatric disorders. Symptoms of HD typically appear between the ages of 30 and 50 and worsen over the next 10 to 25 years, leading to death in approximately 15 years, on average, after the onset of motor signs and symptoms. People with advanced HD need full-time care to help with their day-to-day activities and ultimately succumb to pneumonia, heart failure or other complications. It is estimated that approximately 40,000 people in the United States are affected by HD.
HD is caused by a mutation that leads to an increased number of CAG triplet repeats in Exon 1 of the Huntingtin (HTT) gene. Expression of mutant HTT (mtHTT) negatively affects many cellular functions, leading to neuronal death and brain atrophy as symptoms manifest. Longer CAG repeat lengths (>50) are often associated with juvenile or young adult-onset HD and shorter survival after disease onset.
Wild-type HTT (wtHTT) is thought to be important for normal neuronal function in the adult central nervous system). It is reported to be involved in axonal transport, protein clearance, synaptic function and cell survival. Increasing lines of evidence also suggest that loss of normal HTT function contributes to the HD pathology. Thus, we believe an allele-selective therapeutic that can dial down mtHTT expression and reduce mtHTT mRNA and protein while preserving wtHTT expression represents a highly desirable therapeutic profile.
Our HD program is based on GeneTAC® molecules consisting of a DNA-targeting moiety designed to target the CAG repeats in the Exon 1 region of the HTT gene, linked to a ligand moiety that is designed to dial down the transcription of the mutant allele without disrupting the normal HTT expression. As a result, the HD GeneTAC® small molecules are designed to address the root cause of HD by selectively reducing the toxic mtHTT gene product, apply to a broad spectrum of HD patients, and benefit from the favorable characteristics of small molecule therapeutics that have the potential to distribute to the whole brain and access many cells.
GeneTAC® Platform
Research Programs
Discovery efforts for multiple other small molecule genomic medicines are also underway. We believe our experiences with GeneTAC® molecules allow us to more rapidly design GeneTAC® molecules for additional indications.